An organism’s genome can be thought of as a set of instructions that code for its phenotype: the observable traits which include appearance, behaviour, or illnesses. The expression of specific genes – whether they are turned on or off – is further regulated by a network of DNA sequences and proteins. This regulatory network instructs cells to “read” different parts of the genome, producing a wide range of phenotypes that can be seen on the cellular level in the way that cells develop and function.
Research in the Mitchell Lab studies mechanisms of gene regulation and their effects on stem cells, cell differentiation, and pathology. Our research is focused on three main projects:
Transcriptional Regulatory Mechanisms in Development and Disease
Watch Professor Jennifer Mitchell’s talk on recent research in the Mitchell Lab as part of the Cell and Systems Biology Seminar Series.
We use CRISPR genome editing in our work to make allele specific deletions in F1 mouse cells with a hybrid genome (musculus129/M. castaneus). Read more for a description of this approach and a link to our 129/Cast SNP track for designing CRISPR gRNAs and allele specific primers.
Transcriptional Regulatory Networks in Embryonic and Neural Stem Cells
Pluripotent stem cells have the potential to develop into any cell type in an adult organism, including nerve cells. This pluripotency is maintained by the expression of certain genes. In particular, the SOX2 gene plays a key role in maintaining embryonic and neural stem cells, as well as normal brain development and nerve cell regeneration.
The expression of SOX2 is regulated by specialized DNA sequences called enhancers, which control when and where specific genes are turned on in the brain. As such, mutations in enhancers can cause aberrant expression of SOX2, leading to neurodegeneration, epilepsy and, in severe cases, infant death due to brain malformation.
Our research team has discovered new enhancer sequences which activate the SOX2 gene in stem cells and influence the production of nerve cell precursors from stem cells. We are currently working on determining sequences that are important for the function of these regulatory elements and finding new genes which, like SOX2, are critical for nerve cell regeneration.
Furthermore, our project aims to identify other enhancers that regulate gene expression in the stem cell compartment of the brain and determine how these enhancers are epigenetically modified during brain development and with ageing. This will allow us to better understand why some people develop neurodegenerative diseases such as epilepsy, multiple sclerosis and Huntington’s disease and improve disease diagnosis and treatment using precision medicine approaches.
This project is funded by the Canadian Institutes of Health Research Project Grants Program.
Read more about this work:
Dhaliwal NK, Abatti L, Mitchell JA. KLF4 protein stability regulated by interaction with pluripotency transcription factors overrides transcriptional control. Genes Dev, 2019;33(1): 1-14Dhaliwal NK, Miri K, Davidson S, El Jarkass HT, Mitchell JA KLF4 Nuclear Export Requires ERK Activation and Initiates Exit from Naive Pluripotency Stem Cell Reports 2018;10(4): 1308-1323
Moorthy SD, Davidson S, Shchuka VM, Singh G, Malek-Gilani N, Langroudi L, Martchenko A, So V, Macpherson NN and Mitchell JA. Enhancers and super-enhancers have an equivalent regulatory role in embryonic stem cells through regulation of single or multiple genes. Genome Res. 2017;27: 246-258.
Moorthy SD, Mitchell JA. Generating CRISPR/Cas9 Mediated Monoallelic Deletions to Study Enhancer Function in Mouse Embryonic Stem Cells J Vis Exp 2016, (110), e53552
VM Shchuka, N Malek-Gilani, G Singh, L Langroudi, NK Dhaliwal, SD Moorthy, S Davidson, NN Macpherson and JA Mitchell. Chromatin Dynamics in Lineage Commitment and Cellular Reprogramming. Genes 2015, 6(3), 641-661.
HY Zhou, Y Katsman, NK Dhaliwal, S Davidson, NN Macpherson, M Sakthidevi, F Collura,and JA Mitchell A Sox2 distal enhancer cluster regulates embryonic stem cell differentiation potential. Genes Dev, 2014, 28 (24): 2699-2711.
Decoding the Mammalian Regulatory Genome
Despite the completion of the human genome sequence nearly two decades ago, we are currently unable to determine the function of the vast majority of this sequence. We understand how to read the sequence code (how the 4 bases are translated into amino acids) within gene coding regions, which make up less than 2% of the genome. Yet, we are still limited in our understanding of the non-coding regions that make up the other 98%.
Buried within the non-coding genome are gene regulatory elements that turn genes on or off in specific cells. These regulatory elements play a key role in development as they contain instructions for the development of specific cell types. By activating different groups of genes, regulatory elements can cause stem cells to differentiate into brain, blood, or heart cells, for example.
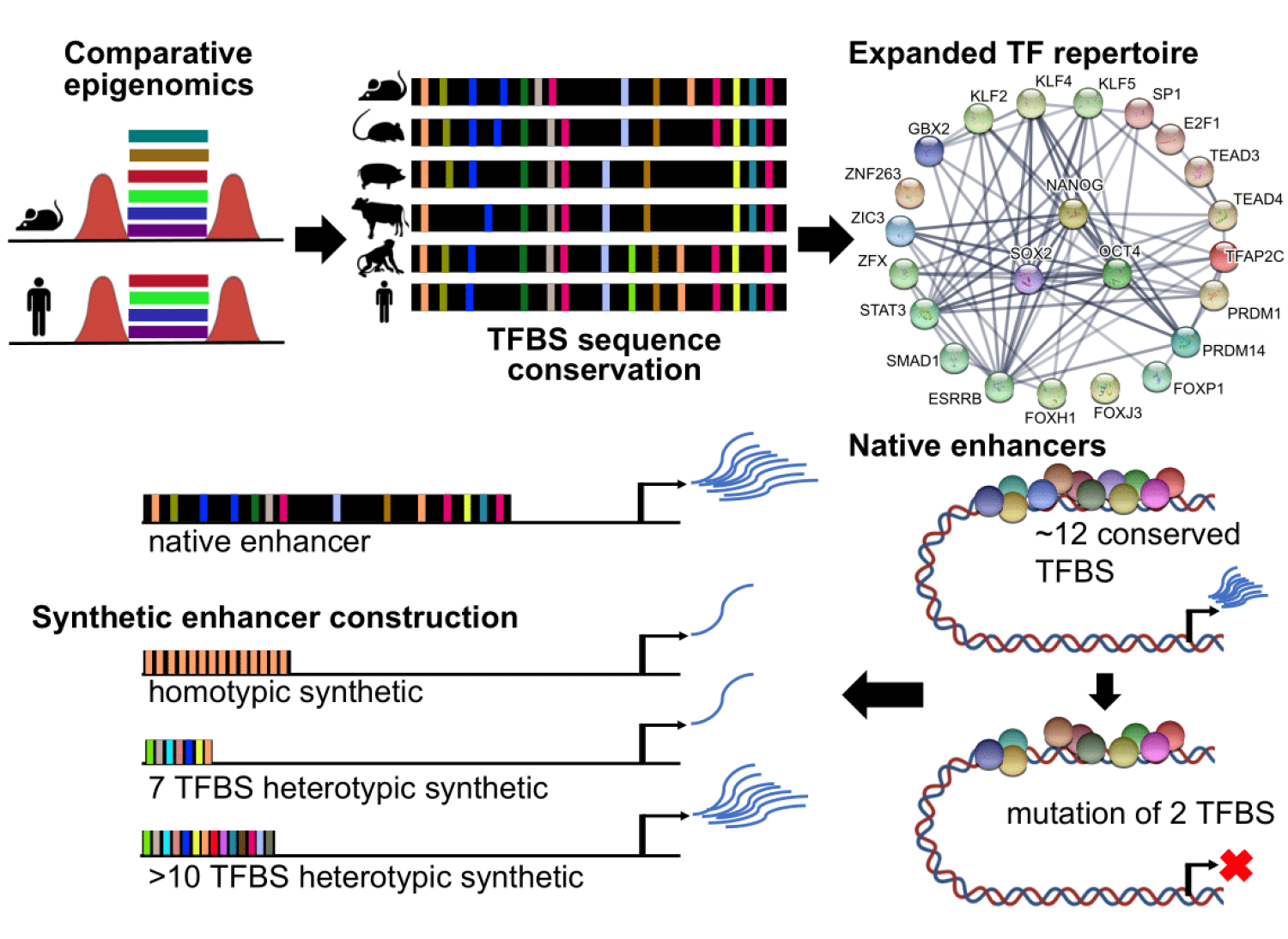
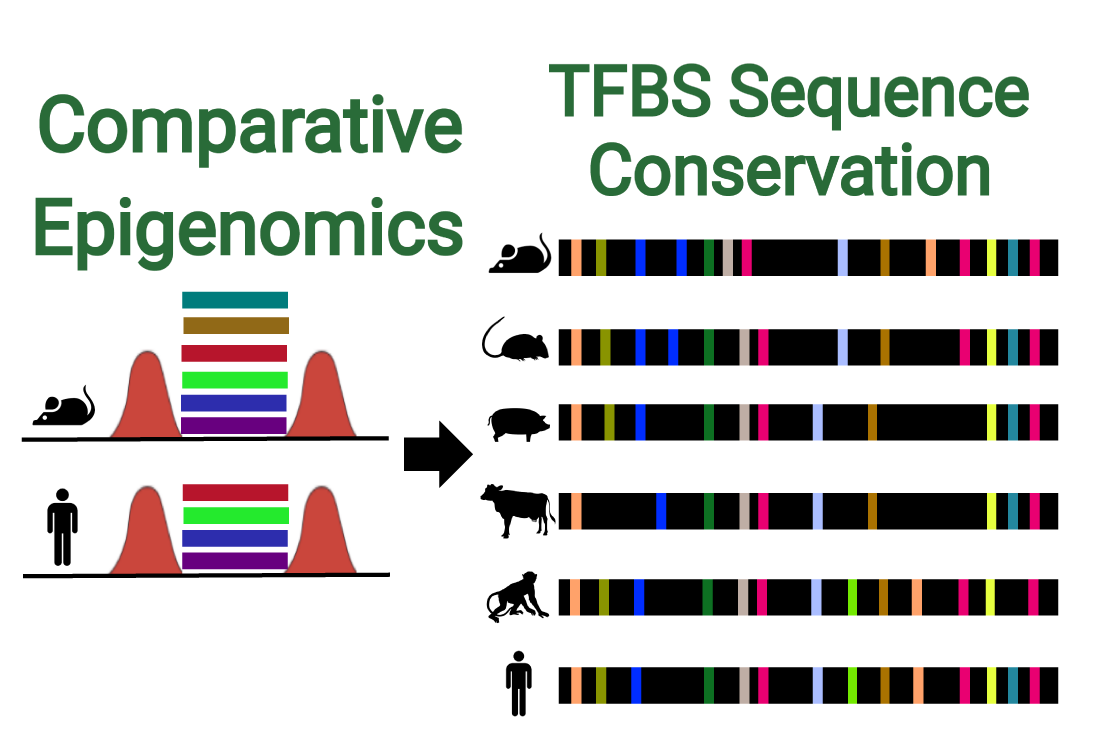
Our research studies a type of regulatory element called enhancers. Enhancers are short DNA sequences of about 500 bases which have an activating effect by turning on specific genes when needed. We know that enhancers are important for the proper development of complex organisms and are often the cause of specific differences in the body plans of different species. However, we know remarkably little about the sequence code that gives enhancers their function.
We are not able to scan the genome sequence to find enhancers or interpret the function of different parts of the sequence when we do know the location of an enhancer. This is like having a book written in a language you can’t read. Or, as we have fully sequenced genomes for more than 100 mammals, an entire library of books that you can’t read.
Recently, our team has determined new information about the enhancer regulatory code. We used computational biology to predict the conserved functional units that are critical to enhancers in pluripotent stem cells. Using sequence conservation, we studied the genomes of embryonic stem cells across six mammals and identified common regions that contain binding sites for transcription factors involved in gene regulation.
In the long term, fully decoding genome sequences will allow us to predict how changes in regulatory sequences will affect the expression of specific genes. This will have widespread impact on our understanding of development, evolution and the underlying cause of phenotype variation in populations.
This research program is funded by the Natural Sciences and Engineering Research Council of Canada Discovery Grants Program.
Read more about this work:
A flexible repertoire of transcription factor binding sites and diversity threshold determines enhancer activity in embryonic stem cells. Singh G, Mullany S, Moorthy SD, Zhang R, Mehdi T, Tian R, Duncan AG, Moses AM, Mitchell JA. doi: https://doi.org/10.1101/2020.04.17.046664
Transcriptional enhancers: from prediction to functional assessment on a genome-wide scale. Tobias IC, Abatti LE, Moorthy SD, Mullany S, Taylor T, Khader N, Filice MA, Mitchell JA.Genome. 2020 Sep 22. doi: 10.1139/gen-2020-0104. Online ahead of print.PMID: 32961076
TF Mehdi, G Singh, JA Mitchell, AM Moses. Variational infinite heterogeneous mixture model for semi-supervised clustering of heart enhancers. Bioinformatics 35 (18), 3232-3239
CY Chen, Q Morris and JA Mitchell. Enhancer identification in mouse embryonic stem cells using integrative modelling of chromatin and genomic features. BMC Genomics, 2012, 13:152.
Discovering the Transcriptional Regulatory Networks that Control the Onset of Preterm Labour
During labour, contractions of the myometrium, a layer of smooth muscle cells in the uterus, generate the force needed to expel the fetus at birth. These contractions are initiated when the expression of specific genes in smooth muscle cells are activated in response to hormonal, mechanical and inflammatory signals.
This cell-coordinated contractile response can occur prior to term, causing women to enter labour too early. In these cases preterm labour can result in the delivery of underdeveloped infants at high risk of death or complications later in life.
Currently, there is no effective treatment for preterm labour, and little is known about how gene expression changes during pregnancy and labour. Recent research has identified activated genes in the myometrium at term and preterm labour, but the mechanisms that stimulate expression of these genes remain unclear. Our team studies the components involved in activating genes in both term labour and preterm labour to better understand the basis of preterm labour and develop new preterm labour therapies.
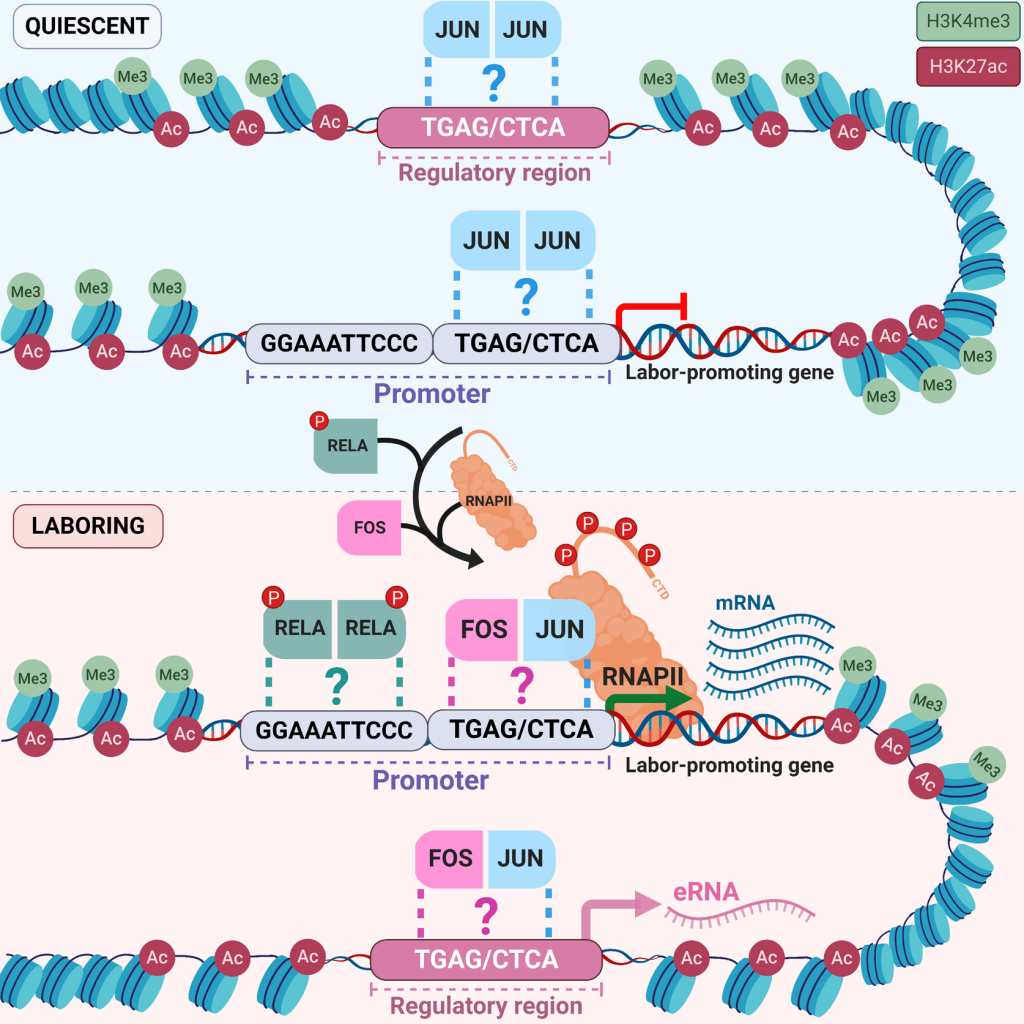
One focus of our research is to identify the genome structures that are specific to myometrial cells and the labour process. The three-dimensional structure in which DNA is organized can influence gene expression by allowing certain regions of the genome to contact genes and turn them on or off. We are also working on identifying the transcriptional regulatory proteins that bind to the genome to regulate gene expression during preterm or term labour.
Uncovering the suite of genomic regions and regulatory proteins that underlie the labour process will provide new targets for the development of therapeutic treatments for preterm labour, preventing complications that occur when babies are born preterm.
This project is funded by the Canadian Institutes of Health Research Project Grants Program.
Read more about this collaborative work:
Transcriptional Control of Parturition: Insights from Gene Regulation Studies in the Myometrium. Khader N, Shchuka VM, Shynlova O, Mitchell JA. doi: https://doi.org/10.1093/molehr/gaab024
Shchuka VM, Abatti LE, Hou H, Khader N, Dorogin A, Wilson MD, Shynlova O, Mitchell JA. The pregnant myometrium is epigenetically activated at contractility-driving gene loci prior to the onset of labor in mice. PLoS Biol 18(7): e3000710. https://doi.org/10.1371/journal.pbio.3000710
JA Mitchell and SJ Lye, Differential Activation of the Connexin 43 Promoter by Dimers of Activator Protein-1 Transcription Factors in Myometrial Cells. Endocrinology, 2005, 146(4): 2048-54.
In order to aid generation and analysis of CRISPR deletions in F1 cells, we have collected all SNPs between the 129 and Cast genomes based on the Sanger Institute Mouse Genomes Project and located them to their coordinates on the mm9 reference genome. Note that SNPs common to both genomes are not marked.
In our paper demonstrating that enhancers and super-enhancers have an equivalent regulatory role in mouse ES cells, we studied the effect of distal regulatory elements on transcription using genome editing. Although reporter gene constructs can provide useful guidance on which elements are active, the best way to examine regulatory element activity is in an endogenous context. One method of doing this is to delete the regulatory elements in the cell using CRISPR/Cas9-mediated deletion.
We used F1 cells of M. musculus129/M. castaneus to study the elements responsible for gene regulation. Given that disruption of one allele of an enhancer could result in changes to transcription in cis, but not in trans, protein levels of the target gene will not be eliminated by monoallelic disruption. Consequently there is less chance of phenotypic changes due to disruption of a regulatory cascade, and we can monitor transcriptional changes in the altered allele without confounding effects.
CRISPR/Cas9-mediated deletion can occur on either allele. To distinguish between alleles in our F1 cells, we have collected all the SNPs between 129 and Cast from the Sanger Institute Mouse Genomes Project and located them to their coordinates on the mm9 reference genome.
These SNPs can be displayed at the UCSC Mouse Genome Browser by following this link. The first base in the 129CastTrack is the 129 SNP, and the second is the Cast SNP. For SNPs at which confidence in the SNP is low, the base is displayed in lower case.
Usage of our SNP track
Determining which allele of the targeted region has been excised
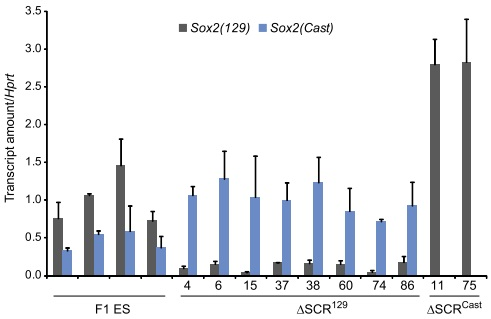
We used CRISPR to target the Sox2 Control Region (SCR) for deletion (Zhou et al, 2014). Guide RNAs targeted the regions designated gRNA104 and gRNA112 in A below. The 129 or Cast alleles were specifically amplified by PCR using the primer pr3’112R1 and allele specific versions of the primer pr5’104F1. As shown for isolate #15 in B, these allele specific primer pairs are used to distinguish the deleted 129 allele from the undeleted Cast allele.

Designing allele specific qPCR primers to determine allelic ratios of transcription
We designed allele specific primers using our SNP database to monitor transcription levels of both alleles of Sox2 in our deleted lines. Quantitative RT-qPCR revealed that in clones lacking the 129 enhancer (ΔSCR129), transcription of the 129 allele of Sox2 was greatly reduced, whereas in clones lacking the Cast enhancer (ΔSCRCast), transcription of the Cast allele of Sox2 was virtually eliminated.